International Journal of Theoretical and Applied Nanotechnology (IJTAN)
ISSN: 1929-1248

Volume 2, Year 2014 - Pages 1-8
DOI: 10.11159/ijtan.2014.001
Application of Quantum Dots to the Study of Liposome Targeting in Leishmaniasis and Malaria
Alba Pujol1, Patricia Urbán2,3,4, Cristina Riera1, Roser Fisa1, Israel Molina5, Fernando Salvador5, Joan Estelrich4,6, Xavier Fernàndez-Busquets2,3,4
1Laboratori de Parasitologia, Departament de Microbiologia i Parasitologia Sanitàries, Facultat de Farmàcia, Universitat de Barcelona, Av. Joan XXIII s/n, E08028 Barcelona, Spain
apujolbrugues@gmail.com, mcriera@ub.edu, rfisa@ub.edu
2Institute for Bioengineering of Catalonia, Baldiri Reixac 10-12, E08028 Barcelona, Spain
3Centre for International Health Research (CRESIB, Hospital Clínic-Universitat de Barcelona), Centre Esther Koplowitz, planta 1, CRESIB, Rosselló 149-153, E08036 Barcelona, Spain
4Institut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, Martí i Franquès 1, E08028 Barcelona, Spain
urban.patricia@gmail.com, xfernandez_busquets@ub.edu
5Infectious Diseases Department, Hospital Universitari Vall d'Hebron, PROSICS Barcelona, Pg. Vall d'Hebron 119-129, E08035 Barcelona, Spain
israelmolina@ymail.com, medinano@yahoo.es
6Departament de Fisicoquímica, Facultat de Farmàcia, Universitat de Barcelona, Av. Joan XXIII s/n, E08028 Barcelona, Spain
joanestelrich@ub.edu
Abstract - Nanotechnological devices for therapeutic applications are massively addressed to diseases prevalent in the developed world, particularly cancer, because of the wrong assumption (for both ethical and technical reasons) that nanomedicines are too expensive and thus they can not be applied to diseases of poverty. Here we have applied quantum dots to study at the cellular level the delivery of the contents of liposomes to erythrocytes infected by the malaria parasite Plasmodium falciparum, and to macrophages infected by the leishmaniasis causative agent Leishmania infantum. A number of works have reported on the encapsulation in liposomes of drugs against both diseases as a strategy to increase therapeutic efficacy and decrease unspecific toxicity. Liposome-carried drugs end up inside Plasmodium-infected red blood cells (pRBCs) and in the phagolysosome system of Leishmania-infected macrophages but some knowledge gaps still obscure subcellular events related to these processes. As a proof of concept, we have used confocal fluorescence microscopy to follow the fate in pRBCs and infected macrophages of quantum dots encapsulated in liposomes, and of lysosomes, leishmaniasis and malaria parasites, nuclei, and phagosomes. Our data indicate that liposomes merge their lipid bilayers with pRBC plasma membranes but are engulfed by macrophages, where they fuse with lysosomes. Lysosomes have not been observed to join with phagosomes harboring single Leishmania parasites, whereas in phagosomes where the parasite has divided there is lysosome-specific fluorescence with a concomitant disappearance of lysosomes from the cytosol. In later stages, all the lysosome-specific label is found inside phagosomes whereas the phagosomal marker cadaverine strongly stains the macrophage nucleus, suggesting that Leishmania infection induces in its later stages nuclear degeneration and, possibly, apoptosis of the host cell. These results indicate that induction of macrophage apoptosis should be explored as a possible strategy used by Leishmania to prepare its egress.
Keywords: Leishmania infantum, leishmaniasis, liposomes, malaria, nanomedicine, nanotechnology, Plasmodium falciparum, quantum dots.
© Copyright 2014 Authors - This is an Open Access article published under the Creative Commons Attribution License terms. Unrestricted use, distribution, and reproduction in any medium are permitted, provided the original work is properly cited.
Date Received: 2013-09-04
Date Accepted: 2013-12-11
Date Published: 2014-01-31
1. Introduction
Leishmania parasites have two morphological forms, termed amastigotes and promastigotes, which are found in the mammalian and sandfly hosts, respectively [1]. Amastigotes are largely intracellular stages that mainly live in the phagolysosomal system of macrophages, the predominant host cell. Macrophages are phagocytic and readily engulf amastigotes, followed by phagosome-lysosome fusion. This microbial defence mechanism is lethal to most foreign organisms, as it results in exposure to an acidic pH of 4.5 to 5.5 and attack by a battery of lysosomal enzymes. However, Leishmania can survive this experience, and indeed thrives in this environment [2]. Parasites within the phagolysosome grow and divide, such that an individual macrophage may eventually contain many tens of amastigotes. There is no specific escape mechanism known, and it is assumed that in most cases the host cell simply ruptures when it cannot accommodate any more parasites, which are then taken up by further macrophages.
The treatment of leishmaniasis will depend on its clinical form, but it is based mainly on pentavalent antimonial compounds. Amphotericin B and its modern liposomal formulation have an important role in current treatment schedules, although high costs limit their use in most endemic countries. New drugs such as miltefosine, the first orally administered treatment against Leishmania, represent attractive alternatives that have been evaluated in several clinical trials [3]. The hidden location of amastigotes inside the macrophage's phagolysosomes is responsible for impairing the accession of therapeutic drugs, rendering these poorly selective, or forcing their administration in repeated and high doses by parenteral routes [4, 5]. These facts contribute to the high toxicity and in most cases to the limited compliance and efficacy of current conventional medication used against all the clinical forms [6, 7, 8]. Drug delivery systems should allow avoiding the adverse effects caused by problematic routes of administration as well as enhancing the antileishmanial activity and reducing the toxicity of the medication [9].
Liposomes, when administered in vivo by a variety of routes, rapidly accumulate in the mononuclear phagocyte system, a phenomenon that can be used to target drugs for the treatment of intracellular parasites that reside in macrophages, such as Leishmania [10]. A number of studies have been conducted to exploit liposomes as drug vehicles for the treatment of leishmaniasis [11-18], but data about the subcellular fate of liposome-encapsulated drugs are scarce.
Current administration methods of antimalarial drugs deliver the free compound in the blood stream, where it can be unspecifically taken up by all cells, and not only by Plasmodium-infected red blood cells (pRBCs). Nanosized carriers have been receiving special attention with the aim of minimizing the side effects of malaria therapy by increasing drug bioavailability and selectivity [19]. To follow the delivery of liposomal contents into pRBCs and Leishmania-infected macrophages we have used luminescent semiconductor nanocrystals termed quantum dots [20]. Quantum dots (QDs) provide unique intrinsic photophysical properties for potential medical, diagnostic and basic research applications, among them high quantum yields and high molar extinction coefficients along with exceptional resistance to both chemical and photodegradation. QD encapsulation into liposomes can be studied by electron microscopy and their release to pRBCs and macrophages can be conveniently followed by fluorescence microscopy over a wide concentration range, a versatility that can not be matched by small fluorochromes.
2. Materials and Methods
Except where otherwise indicated, all reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.1. Liposome Formation
Liposomes were prepared by the lipid film hydration method [21]. Different lipid combinations were tested in order to establish a liposomal formulation with low hemolytic activity and low general cytotoxicity. Lipids (1,2-dioleoyl-sn-glycero-3-phosphatidylcholine, DOPC; 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl) butyramide], MPB-PE; Avanti Polar Lipids Inc., Alabaster, AL, USA; both ≥ 99% purity according to thin layer chromatography analysis) were dissolved in chloroform:methanol (2:1 v/v) in a round-bottomed flask. Organic solvents were removed by rotary evaporation under reduced pressure at 37 °C to yield a thin lipid film on the walls of the flask, and remaining solvent traces were eliminated by drying under N2 flow for 30 min. Films were left overnight in a desiccator to ensure the complete removal of chloroform. The dry lipids were hydrated in phosphate-buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4) at 37 °C to obtain a concentration of 10 mM and multilamellar liposomes were formed by 3 cycles of constant vortexing followed by bath sonication for 4 min each. Multilamellar liposomes were downsized to form uni- or oligolamellar vesicles by extrusion through 200-nm polycarbonate membranes (Poretics, Livermore, CA, USA) in an extruder device (LiposoFast; Avestin, Ottawa, Canada). Liposome size was determined by dynamic light scattering using a Zetasizer NanoZS90 (Malvern Ltd, Malvern, UK). Liposomes encapsulating 120 nM 655 ITK™ carboxyl quantum dots (Molecular Probes, Eugene, OR, USA) were prepared by dissolving QDs in the hydration buffer. At the concentrations used in macrophage and pRBC cultures, QDs were neither cytotoxic nor hemolytic. Immunoliposomes bearing on their surfaces specific antibodies against pRBCs were prepared following established protocols [22]. Liposomes were pelleted by ultracentrifugation (100,000× g, 2×, 45 min, 4 °C), and finally resuspended in 10 volumes of PBS and kept at 4 °C for up to 2 weeks before adding them to cultures.
2.2. Cryogenic Transmission Electron Microscopy (cryo-TEM)
For cryo-TEM analysis of the preparations of liposomes loaded with quantum dots, a thin aqueous film was formed by placing a 5-µl sample drop of the liposome suspension on a glow-discharged holey carbon grid and then blotting the grid against filter paper. The resulting thin sample films spanning the grid holes were vitrified by plunging the grid (kept at 100% humidity and room temperature) into ethane, which was maintained at its melting point with liquid nitrogen, using a Vitrobot (FEI Company, Eindhoven, The Netherlands). The vitreous films were transferred to a Tecnai F20 TEM (FEI Company)using a Gatan cryotransfer (Gatan, Pleasanton, CA), and the samples were observed in a low dose mode. Images were acquired at 200 kV at a temperature between –170 and –175 °C and low-dose imaging conditions below 20 e–/Å2, with a 4096×4096 pixel CCD Eagle camera (FEI Company).
2.3. Assay on Intracellular Leishmania infantum Amastigotes
Promastigotes of the MCAN/ES/92/BCN503 L. infantum strain were cultured at 26 °C in Schneider's medium, pH 7.0, supplemented with 20% heat-inactivated foetal bovine serum (FBS; SLI Barcelona Antibody), 25 μg/ml gentamycin and 1% penicillin (100 U/ml)–streptomycin (100 μg/ml), and were collected during stationary phase at a concentration of 1×108 cells/ml. Subsequently, the parasites were labelled with 2.5 µg/ml carboxyfluorescein diacetate N-succinimidyl esther (CFSE) solution in PBS, for 5 min at 26 °C. Labelling was quenched with an equal volume of supplemented Schneider's culture medium. Peritoneal macrophages were obtained after stimulation of female Swiss mice with 3 ml of 3% sodium thioglycolate. After 48 h, 4 ml of physiological serum at 4 °C was injected intraperitoneally, and after 15 min peritoneal fluid was collected and centrifuged. Suspensions of 5×104 cells/ml in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin (100 U/ml)–streptomycin (100 μg/ml) solution were prepared and 0.3 ml was seeded in a LabTek 8 chamber slide system (Nalge Nunc International, Rochester, NY, USA) and incubated at 37 °C for 24 h in a 5% CO2 atmosphere. After incubation the medium was removed and 0.3 ml of late stationary phase CFSE-stained promastigotes was added at a concentration of 5×106 cells/ml. After 4 h of incubation at 35 °C, extracellular parasites were removed by washing with sterile PBS, and 0.2 ml of liposome-encapsulated QD suspension in PBS was mixed with 0.2 ml of RPMI-1640 medium and the resulting 0.4 ml (containing 60 nM QDs) was added to each well and incubated at 37 °C in a 5% CO2 atmosphere for the times indicated. The cells were subsequently washed and slides were prepared for confocal fluorescence microscopy.
2.4. Plasmodium falciparum Cell Culture
The P. falciparum 3D7 strain was grown in vitro in group B human RBCs using previously described conditions [23]. Treatment of cultures with QD-containing liposomes was done following established protocols [22].
2.5. Cytotoxicity and Hemolysis Assays
For cytotoxicity assays, serial dilutions of QD solution in RPMI-1640 medium were added to 100 μl of the murine monocyte-macrophage cell line Raw 264.7 cultured in 96-well plates (Costar Corning Inc.) at a concentration of 5×104 cells/well. After incubation for 24 h at 37 °C in a 5% CO2 atmosphere, 10 μl of 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate labelling reagent (WST-1; Roche Diagnostics GmbH) was added to each well, and the plate was incubated in the same conditions for 4 h. After thoroughly mixing for 1 min on a shaker, the absorbance of the samples was measured at 450 nm using a Titerek Multiskan Plus MK II 314 (Bio-Rad Laboratories Inc.). WST-1 in the absence of cells was used as blank and samples were prepared in triplicate for each experiment. Hemolysis assays were performed as described elsewhere [22].
2.6. Confocal Microscopy
Macrophage and Leishmania nuclei were stained for 10 min with either 1 μg/ml Hoechst 33342 (Molecular Probes) or 1 μM TOPRO-3 (Invitrogen), lysosomes for 30 min with 50 nM Lyso Tracker Red (DND-99; Molecular Probes), and phagosomes for 10 min with 0.05 nM mono dansyl cadaverine. RBC membranes, Plasmodium nuclei, and secondary antibodies were stained following established protocols [22]. After the corresponding incubations and PBS washing steps, the samples were fixed for 20 min with 3% (v/v) paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4, and finally mounted with Prolong Gold (Invitrogen) following standard protocols. Samples were imaged with a Leica TCS SP5 laser scanning confocal microscope equipped with an acoustic optical beam splitter, a DMI6000 inverted microscope, blue diode (405 nm), Ar (458/476/488/496/514 nm), diode pumped solid state (561 nm), and HeNe (594/633 nm) lasers, and APO 63× oil (NA 1.4) or glycerol (NA 1.3) immersion objective lenses. Hoechst 33342 (or 4',6-diamino-2-phenylindole, DAPI), quantum dot, CFSE (or fluorescein-labelled secondary antibody), DND-99 (or WGA-rhodamine), TOPRO-3, and mono dansyl cadaverine images were acquired sequentially using 405, 458, 488, 561, 633 and 405 nm laser lines, and emission detection ranges 415–480, 643–735, 500–545, 571–630, 643–735 and 500–545 nm, respectively, with the confocal pinhole set at 1 Airy units. Images were acquired at 400 Hz in a 512×512 pixels format, 8× zoom, and pixel size of 60×60 nm.
3. Results
According to cryo-TEM analysis, 655 ITK™ carboxyl QD-loaded liposomes contained variable numbers of QDs that were either associated to the membrane or free in the liposomal lumen (Figure 1). Under the preparation conditions used in this work, approximately half of the liposomes contained at least one QD. QDs were observed to be ca. 5×10 nm rods, in agreement with the maker's information (Web-1). They have a CdSe core encapsulated in a crystalline shell of ZnS and an external amphiphilic polymer coating to prevent formation of free Cd, and are widely used in live-cell in vitro assays (Web-1).
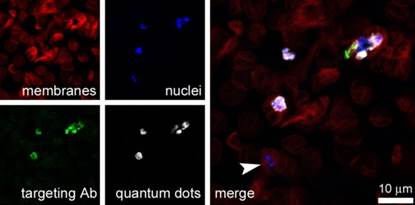
QDs have been explored as an aid to characterize the targeting of immunoliposomes designed for the delivery of antimalarial drugs to pRBCs. A prototype nanovector was constructed consisting of 120 nM 655 ITK™ carboxyl QDs encapsulated in immunoliposomes with the formulation DOPC:cholesterol:MPB-PE 77.5:20:2.5, which carried a specific antibody against pRBCs. QD-loaded immunoliposomes were added to 3% hematocrit living cultures of the P. falciparum 3D7 strain at a concentration in the dish of 1 mM lipid and 60 nM QD to analyze whether they exhibited the expected targeting towards pRBCs. At this concentration QD-containing liposomes did not induce detectable hemolysis. After incubating for 90 min with gentle stirring the samples were processed for detection by confocal fluorescence microscopy of cell membranes, QDs, targeting antibody, and parasite nuclei. The results obtained showed an invariable colocalization of the four signals only in late form-containing pRBCs, but not in the early forms also called ring stages (Figure 2). QD signal in pRBCs was scattered throughout the cell, without evidence of containment inside any particular organelles or vesicular structures. No intracellular QD fluorescence was observed when free 60 nM QDs were added to cultures (data not shown).
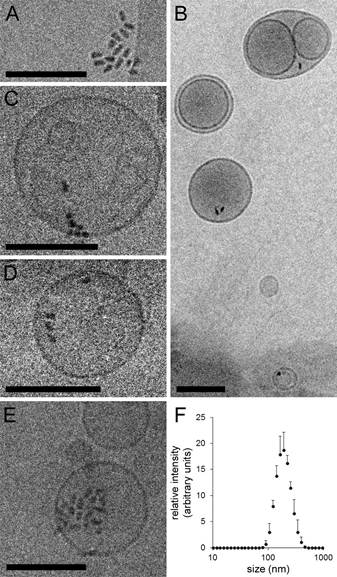
Figure 1. Encapsulation of QDs in liposomes. (A) Cryo-TEM image of 655 ITKTM carboxyl QDs. (B-E) Cryo-TEM images of the QD-containing liposomes obtained with the protocol described in the Methods. (F) Typical dynamic light scattering plot of a QD-containing liposome sample, with a mean diameter of 179 nm and a mean polydispersity index of 0.08. Scale bar: 100 nm.
In a preliminary approach to the application of QD-loaded liposomes to leishmaniasis research, we analyzed by fluorescence confocal microscopy the subcellular localization of QDs encapsulated in DOPC:cholesterol 80:20 liposomes that were delivered to Leishmania-infected macrophages. At their final concentration present in the cultures (60 nM), QDs did not induce detectable cytotoxicity in macrophage assays (Figure 3).
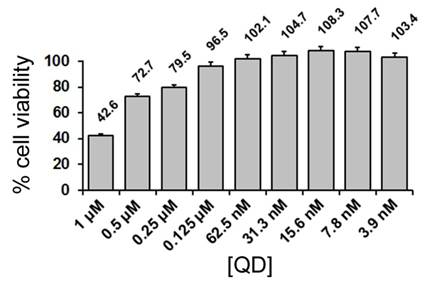
Figure 3. Cell viability assay of the effect of QDs on macrophages.
One hour after the addition of the liposome suspension to a macrophage culture the QD fluorescence could be already observed inside the cells as whole internalized liposomes fused with lysosomes (Figure 4A). Figure 1B shows a macrophage 27 h post-infection with three phagosomes (arrowheads), or parasitophorous vacuoles, in whose interior the green fluorescence corresponding to the prior labelling of Leishmania parasites with CFSE can be observed. The punctate pink fluorescence corresponds to lysosomes labelled with the lysosome tracker DND-99. Colocalization analysis of QDs and lysosomes confirmed that internalized liposomes fuse with lysosomes (Figure 4C). Because of the lack of significant QD fluorescence not colocalizing with lysosomes, we conclude that any fraction of QDs not encapsulated in liposomes will also end up in the lysosome system, in agreement with previous data [24]. The arrowhead in the merge panel shows the parasitophorous vacuole, which does not contain QDs. In the early stages of infection, up to 27 h, single parasites could be observed inside the phagosome, and numerous lysosomes were found scattered throughout the macrophage cytosol (Figure 4D). In Figure 4E is shown a macrophage in a more advanced stage of infection containing two vacuoles, one probably with a single parasite (arrowhead), and a second vacuole where Leishmania had replicated, as suggested by lighter CFSE staining of the individual amastigotes (arrow). Here, lysosomes were essentially absent from the cytosol and their specific fluorescence was strongly localized inside the vacuole where Leishmania had already divided, but not in the vacuole containing only one parasite. In later stages, 52 h post-infection, we have observed strong staining of the macrophage nucleus with mono dansyl-cadaverine (Figure 4F), a phagosome marker. As in Figure 4C, also corresponding to a sample 52 h post-infection, the macrophage nucleus appeared swollen.
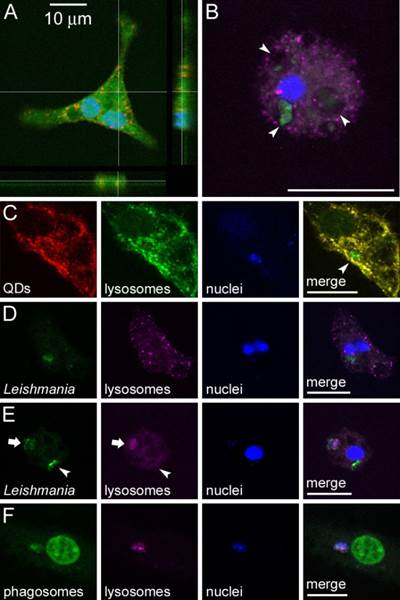
Figure 4. Confocal fluorescence analysis of L. infantum-infected macrophages treated with QD-containing liposomes. (A) Non-infected macrophage showing QD fluorescence in red 1 h after addition of QD-loaded liposomes to the cell culture; the macrophage nucleus and cytosol have been stained with Hoechst and CFSE, respectively. (B) A macrophage stained with Hoechst (nucleus, blue), DND-99 (lysosomes, purple), and CFSE (Leishmania, green), 27 h after infection. The arrowheads indicate three phagosomes. (C) Colocalization analysis of QDs that had been administered encapsulated in liposomes 1 h before, of lysosomes, and of Leishmania nuclei stained with TOPRO-3, 52 h after infection. For an easier interpretation of the merge panel, lysosomes are here shown in green color. (D,E) Colocalization analysis of Leishmania, lysosomes, and macrophage nuclei, 27 h after infection. (F) Colocalization analysis of cadaverine (a phagosomal marker also staining here the macrophage's nucleus), lysosomes, and TOPRO-3-stained parasite nuclei, 52 h after infection. Scale bar: 10 µm.
Likely, the progression of infection was at an earlier stage in panel C, as suggested by abundant lysosomes still present in the cytosol.
4. Discussion
In leishmaniasis, the interactions of phagosomes harboring L. donovani promastigotes with late endosomes and lysosomes are inhibited until the promastigote has fully differentiated into an amastigote [25]. Lipophosphoglycan, the major surface glycoconjugate on L. donovani promastigotes, is transferred from the parasite to the host macrophage membrane during phagocytosis and induces periphagosomal actin accumulation correlating with an inhibition of phagosomal maturation [26]. The resulting increased membrane rigidity has been shown to reduce phago-lysosomal fusion [27], suggesting that alteration of the biophysical properties of membranes may be the reason for the reduced ability of the host macrophages to accomplish phagosomal maturation upon infection.
In the nuclei of macrophages that had been infected by L. infantum ca. 52 h earlier, we have observed signs compatible with apoptosis, such as cadaverine-positive labelling and nuclear swelling. Because these symptoms of nuclear degeneration are coincident with the disappearance of lysosomes from the cytosol, it is likely that the phagosome-lysosome fusion triggers alterations in the macrophage nucleus. This in turn suggests that a signal should travel from the phagosome to the nucleus to start the process. Since we have observed that phago-lysosomal fusion proceeds only after intraphagosomal replication of L. infantum, some event related to the promastigote-amastigote transition might be ultimately responsible for the release of the predicted chemical signal(s). Although it has been reported that Leishmania infection protects macrophages from exogenously-induced apoptosis, the corresponding data were obtained 16 h [28] and 24 h post-infection [29]. Our results suggest that at longer times the parasite might be inducing macrophage apoptosis, perhaps as a strategy to prepare its egress. The quantitative incorporation of liposomal contents (QDs) into cytosolic lysosomes and the subsequent phagosome-lysosome fusion, show that lysosomes can target liposomal anti-Leishmania drugs towards the parasite-containing phagosomes. The data presented here indicate that liposome-lysosome fusion is then a prior event to phagolysosome formation and this knowledge can be of use for the design of new targeted drug delivery strategies against leishmaniasis. In the case of malaria,
In the case of malaria, the homogeneous distribution inside pRBCs of QDs delivered within liposomes suggests that the liposomal contents enter the cell through a process of fusion of the liposome lipid bilayer with the cell plasma membrane. RBCs have poor endocytic processes, and for this reason liposomes docked by specific antibodies can be an efficient system to deliver drugs into the cell by such membrane fusion process [30], which occurs on a time scale of milliseconds [31].The remarkable capacity of liposomes to inject their contents into pRBCs presumably has its basis on alterations of the pRBC plasma membrane rendering it less elastic [32], and thus limiting the rebounding of colliding liposomes. The resulting slightly longer interactions, a phenomenon exacerbated if targeting molecules are present, likely allow enough time for the physical phenomenon of lipid bilayer fusion to occur.
Finally, since QDs have a size similar to that of a large protein, the data presented here indicate that immunoliposomes can be a valid system for the delivery to pRBCs of possible future macromolecular antimalarial drugs.
5. Conclusion
Liposomes encapsulating QDs have provided valuable information regarding the subcellular localization of their contents once target cells are reached, i.e. the Plasmodium-infected erythrocyte and the Leishmania-parasitized macrophage (Figure 5). This knowledge can be of use for the design of new therapies specifically targeting particular molecular components of the cell. The above results highlight the potential of nanomedicine for the treatment of infectious diseases of poverty, and advocate for a determined entry of nanotechnology in the fight against widespread pathologies in low per capita income regions.
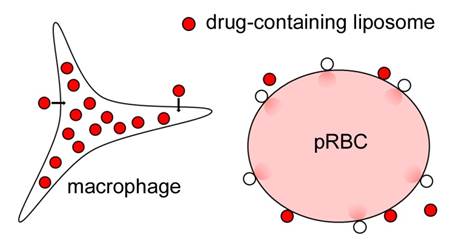
Figure 5. Cartoon depicting the different fates of liposome contents upon interaction with either Leishmania-infected macrophages or pRBCs, which result respectively in liposome internalization and subsequent fusion with lysosomes, or in fusion with the pRBC plasma membrane and injection of liposome cargo into the cytosol.
Acknowledgements
This work was supported by grants BIO2008-01184 and BIO2011-25039, from the Ministerio de Economía y Competitividad, Spain, which included FEDER funds, and by grant 2009SGR-760 from the Generalitat de Catalunya, Spain. We acknowledge the support of the Confocal Microscopy and Cryo-Electron Microscopy Units at the Scientific and Technological Centres from the University of Barcelona (CCiT-UB).
References
[1] P.A. Bates, M.E. Rogers "New insights into the developmental biology and transmission mechanisms of Leishmania" 2004, Current Molecular Medicine, 4, 601-609. View Article
[2] D. Zilberstein, M. Shapira "The role of pH and temperature in the development of Leishmania parasites" 1994, Annual Review of Microbiology, 48, 449-470. View Article
[3] C.R. Davies, P. Kaye, S.L. Croft, S. Sundar "Leishmaniasis: new approaches to disease control" 2003, British Medical Journal, 326, 377-382. View Article
[4] H.W. Murray, J.D. Berman, C.R. Davies, N.G. Saravia "Advances in leishmaniasis" 2005, Lancet, 366, 1561-1577. View Article
[5] S. Sundar, M. Rai "Treatment of visceral leishmaniasis" 2005, Expert Opinion on Pharmacotherapy, 6, 2821-2829. View Article
[6] S. Sundar, T.K. Jha, C.P. Thakur, J. Engel, H. Sindermann, C. Fischer, K. Junge, A. Bryceson, J. Berman "Oral miltefosine for Indian visceral leishmaniasis" 2002, New England Journal of Medicine, 347, 1739-1746. View Article
[7] S. Sundar, H. Mehta, A.V. Suresh, S.P. Singh, M. Rai, H.W. Murray "Amphotericin B treatment for Indian visceral leishmaniasis: conventional versus lipid formulations" 2004, Clinical Infectious Diseases, 38, 377-383. View Article
[8] C.P. Thakur, S. Narayan "A comparative evaluation of amphotericin B and sodium antimony gluconate, as first-line drugs in the treatment of Indian visceral leishmaniasis" 2004, Annals of Tropical Medicine and Parasitology, 98, 129-138. View Article
[9] E.L. Romero, M.J. Morilla "Drug delivery systems against leishmaniasis? Still an open question" 2008, Expert Opinion on Drug Delivery, 5, 805-823. View Article
[10] A.K. Agrawal, C.M. Gupta "Tuftsin-bearing liposomes in treatment of macrophage-based infections" 2000, Advanced Drug Delivery Reviews, 41, 135-146. View Article
[11] R.R. Ribeiro, E.P. Moura, V.M. Pimentel, W.M. Sampaio, S.M. Silva, D.A. Schettini, C.F. Alves, F.A. Melo, W.L. Tafuri, C. Demicheli, M.N. Melo, F. Frezard, M.S. Michalick "Reduced tissue parasitic load and infectivity to sand flies in dogs naturally infected by Leishmania (Leishmania) chagasi following treatment with a liposome formulation of meglumine antimoniate" 2008, Antimicrobial Agents and Chemotherapy, 52, 2564-2572. View Article
[12] D.A. Schettini, A.P. Costa Val, L.F. Souza, C. Demicheli, O.G. Rocha, M.N. Melo, M.S. Michalick, F. Frezard "Pharmacokinetic and parasitological evaluation of the bone marrow of dogs with visceral leishmaniasis submitted to multiple dose treatment with liposome-encapsulated meglumine antimoniate" 2005, Brazilian Journal of Medical and Biological Research, 38, 1879-1883. View Article
[13] A.G. Tempone, D. Perez, S. Rath, A.L. Vilarinho, R.A. Mortara, H.F. de Andrade, Jr. "Targeting Leishmania (L.) chagasi amastigotes through macrophage scavenger receptors: the use of drugs entrapped in liposomes containing phosphatidylserine" 2004, Journal of Antimicrobial Chemotherapy, 54, 60-68. View Article
[14] J.E. Valladares, J. Freixas, J. Alberola, C. Franquelo, C. Cristofol, M. Arboix "Pharmacokinetics of liposome-encapsulated meglumine antimonate after intramuscular and subcutaneous administration in dogs" 1997, The American Journal of Tropical Medicine and Hygiene, 57, 403-406. View Article
[15] N.A. Kshirsagar, P.C. Gokhale, S.K. Pandya "Liposomes as drug delivery system in leishmaniasis" 1995, Journal of the Association of Physicians of India, 43, 46-48. View Article
[16] G. Banerjee, S. Medda, M.K. Basu "A novel peptide-grafted liposomal delivery system targeted to macrophages" 1998, Antimicrobial Agents and Chemotherapy, 42, 348-351. View Article
[17] A.J. Coukell, R.N. Brogden "Liposomal amphotericin B. Therapeutic use in the management of fungal infections and visceral leishmaniasis" 1998, Drugs, 55, 585-612. View Article
[18] C.R. Alving "Liposomes as drug carriers in leishmaniasis and malaria" 1986, Parasitology Today, 2, 101-107. View Article
[19] P. Urbán, X. Fernàndez-Busquets "Nanomedicine against malaria" 2014, Current Medicinal Chemistry, 21, 605-629. View Article
[20] J.B. Delehanty, H. Mattoussi, I.L. Medintz "Delivering quantum dots into cells: strategies, progress and remaining issues" 2009, Analytical and Bioanalytical Chemistry, 393, 1091-1105. View Article
[21] R.C. MacDonald, R.I. MacDonald, B.P. Menco, K. Takeshita, N.K. Subbarao, L.R. Hu "Small-volume extrusion apparatus for preparation of large, unilamellar vesicles" 1991, Biochimica et Biophysica Acta (BBA) - Biomembranes, 1061, 297-303.View Article
[22] P. Urbán, J. Estelrich, A. Cortés, X. Fernàndez-Busquets "A nanovector with complete discrimination for targeted delivery to Plasmodium falciparum-infected versus non-infected red blood cells in vitro" 2011, Journal of Controlled Release, 151, 202-211. View Article
[23] S.L. Cranmer, C. Magowan, J. Liang, R.L. Coppel, B.M. Cooke "An alternative to serum for cultivation of Plasmodium falciparum in vitro" 1997, Transactions of the Royal Society of Tropical Medicine and Hygiene, 91, 363-365. View Article
[24] L.W. Zhang, N.A. Monteiro-Riviere "Mechanisms of quantum dot nanoparticle cellular uptake" 2009, Toxicological Science, 110, 138-155. View Article
[25] R. Lodge, A. Descoteaux "Modulation of phagolysosome biogenesis by the lipophosphoglycan of Leishmania" 2005, Clinical Immunology, 114, 256-265. View Article
[26] M.E. Winberg, A. Holm, E. Sarndahl, A.F. Vinet, A. Descoteaux, K.E. Magnusson, B. Rasmusson, M. Lerm "Leishmania donovani lipophosphoglycan inhibits phagosomal maturation via action on membrane rafts" 2009, Microbes Infection, 11, 215-222. View Article
[27] E. Hayakawa, F. Tokumasu, G.A. Nardone, A.J. Jin, V.A. Hackley, J.A. Dvorak "A Mycobacterium tuberculosis-derived lipid inhibits membrane fusion by modulating lipid membrane domains" 2007, Biophysical Journal, 93, 4018-4030. View Article
[28] M.J. Donovan, B.Z. Maciuba, C.E. Mahan, M.A. McDowell "Leishmania infection inhibits cycloheximide-induced macrophage apoptosis in a strain-dependent manner" 2009, Experimental Parasitology, 123, 58-64. View Article
[29] K.J. Moore, G. Matlashewski "Intracellular infection by Leishmania donovani inhibits macrophage apoptosis" 1994, Journal of Immunology, 152, 2930-2937. View Article
[30] P. Urbán, J. Estelrich, A. Adeva, A. Cortés, X. Fernàndez-Busquets "Study of the efficacy of antimalarial drugs delivered inside targeted immunoliposomal nanovectors" 2011, Nanoscale Research Letters, 6, 620. View Article
[31] J.C. Shillcock, R. Lipowsky "Tension-induced fusion of bilayer membranes and vesicles" 2005, Nature Materials, 4, 225-228. View Article
[32] Q. Chen, M. Schlichtherle, M. Wahlgren "Molecular aspects of severe malaria" 2000, Clinical Microbiology Reviews, 13, 439-450. View Article
Website References
Qdot® ITK™ Carboxyl Quantum Dots, consulted 25 November 2013. View Website