International Journal of Theoretical and Applied Nanotechnology (IJTAN)
ISSN: 1929-1248

Volume 1, Issue 1, Year 2012 - Pages 99-104
DOI: 10.11159/ijtan.2012.015
Laser Fragmentation as an Efficient Size-Reduction Method for Pulmonary Drug Discovery: Proof-of-Concept Study of Beclomethasone Dipropionate
Weimeng Ding¹, Jean-Philippe Sylvestre¹, Grégoire Leclair², Michel Meunier¹
¹Canada Research Chair in Laser Micro/nano-engineering of materials, Laser Processing and Plasmonics Laboratories, Department of Engineering Physics, École Polytechnique de Montréal, P.O. Box 6079, Downtown Station, Montréal, Québec, H3C 3A7, Canada
weimeng.ding@polymtl.ca; jean-philippe.sylvestre@polymtl.ca; michel.meunier@polymtl.ca
²Faculty of Pharmacy, Université de Montréal, P.O. Box 6128, Downtown Station, Montréal, Québec, H3C 3J7, Canada;
gregoire.leclair@umontreal.ca
Abstract - Femto- and nanosecond lasers were used to produce micron-sized particles of beclomethasone dipropionate (BDP) in water. These novel processes were compared with traditional milling methods in terms of particle size, degradation and physicochemical properties. Results showed that similarly to conventional milling methods, minimal chemical degradation and amorphization were induced by the laser processes. As laser micronization requires minimal amounts of drug, this process is well suited for the discovery of pulmonary drugs.
Keywords: Laser Fragmentation, Particle-Size Reduction, Pulmonary Drug, Drug Discovery, Characterization of Organic Materials
© Copyright 2012 Authors - This is an Open Access article published under the Creative Commons Attribution License terms. Unrestricted use, distribution, and reproduction in any medium are permitted, provided the original work is properly cited.
1. Introduction
Since its first introduction (Patil et al., 1987), laser fragmentation in liquid has become a well-established technique to produce nanoparticles (Besner and Meunier, 2010). However, it has been mainly used to produce inorganic nanoparticles, such as metals, alloys and semiconductors. In contrast, organic materials degrade under milder conditions and their particle size reduction is more challenging. So far, only a few organic nanoparticles have been produced by laser fragmentation (Tamaki et al., 2002; Jeon et al., 2007; Hobley et al., 2007; Asahi et al., 2008; Wagener and Barcikowski, 2010). Even fewer works on laser-fabricated drug nanoparticles, including our previous studies on paclitaxel (Kenth et al., 2011) and megestrol acetate (Sylvestre et al., 2011), have been reported.
Laser fragmentation presents an advantage for drug discovery. In the pharmaceutical industry, reducing the particle size has long been a common practice for poorly water-soluble drugs. Smaller drug particles have a higher surface/volume ratio, which increases their dissolution kinetics and improves their absorption in the body (Mosharraf and Nyström, 1995; Rasenack et al., 2003). Several particle size-reduction methods, such as milling and high-pressure homogenisation (Rabinow, 2004; Merisko-Liversidge et al., 2003; Keck and Müller, 2006), are mainly used for industrial production up to hundreds of kilograms. However, these methods are not applicable for drug discovery, as drug candidates at an early stage are available only in milligram quantities. Complementarily, laser fragmentation can be operated on small amount of drugs and is well adapted for drug discovery.
As a tool for reducing the particle size of drugs, laser fragmentation also presents some limitations. The studies of Kenth et al. (2011) and Sylvestre et al. (2011) showed that both micron-sized (1 to 10 µm) and submicron-sized (400 to 900 nm) drug particles were successfully produced by laser ablation or fragmentation. However, submicron-sized particles were more degraded (about 10%) than microparticles (about 2%). From a pharmaceutical point of view, degradation should be as low as possible to maximize safety and efficacy. Consequently, laser fragmentation seems suitable for drug micronization, while its applications for drug nanonization appear to be limited by degradation issues.
While most poorly water-soluble oral and parenteral drugs are best absorbed as nanoparticles, it is different for pulmonary drugs. Because of the branch-like structure of the lungs, particles between 0.5 and 5 µm are well deposited and absorbed in the lungs. Particles bigger than 5 µm cannot penetrate deeply in the lungs and are eliminated by the mucocilary clearance. Particle smaller than 0.5 µm are easily exhaled (Taylor, 2007) and poorly absorbed. Thus, it is of particular interest to evaluate laser fragmentation on a pulmonary drug. In this paper, we used beclomethasone dipropionate (BDP) as a pulmonary drug model. We investigated several laser conditions in order to produce BDP particles in the 0.5 to 5 µm range, evaluated the physicochemical properties of the laser-processed drug, and compared laser technique with the standard milling methods.
2. Materials and Methods
2. 1. Materials
Beclomethasone Dipropionate (BDP) (> 99%) was purchased from Sigma-Aldrich (Oakville, Canada). Methanol (HPLC grade) used for HPLC analyses was purchased from Fisher Scientific (Ottawa, Canada). All aqueous solutions were prepared with deionised water (18.2 MW×cm) generated with a Millipore Milli-Q system (Bedford, MA).
2. 2. Laser Fragmentation
Suspensions of BDP were prepared at concentrations of 0.5 and 2 mg/mL in 2 or 10 mL of deionised water. After 30 min of sonication, laser fragmentation was performed by focusing a laser radiation on a magnetically stirred suspension. Femtosecond laser (120 fs, 800 nm, 1 kHz, Ti:sapphire Hurricane laser, Newport Corporation, power range: 50 to 400 mW) and nanosecond laser (5 ns, 1064 nm, 10 Hz, Nd:YAG Brilliant B laser, Quantel, power range: 1.5-5 W) were used. In this study, these two lasers were chosen in order to coherently compare results with the previous studies (Kenth et al., 2011 and Sylvestre et al., 2011). According to the results in these studies, fs laser offers more versatile conditions than ns laser, but the latter is financially more accessible. To process the drug suspensions, the laser beam (fs or ns) was focused by a lens with a focal length of 10 cm. The irradiation duration varied from 0.5 to 20 h. BDP particles were fragmented by varying the following parameters: suspension concentration and volume, laser power and radiation duration. Experiments were triplicated.
2. 3. Particle Size and Morphology
The average particles size of BDP was first measured by dynamic light scattering (DLS) (Zetasizer NS, Malvern) as a rapid screening method. BDP microsuspensions were then produced at selected conditions, and the more accurate particle size distribution was then measured by Laser Diffraction (LD) (LA-950 + MiniFlow, Horiba Instruments). Dispersant was water and a complex refractive index of 1.55 ‑ 0.1 i was utilized. For scanning electron microscopy analyses (SEM), a drop of the suspension was first dried on a silicon substrate under vacuum, and then observed using a field-emission scanning electron microscope (FE-SEM S-4700, Hitachi).
2. 4. Degradation and Content
Drug purity and degradation was evaluated with high performance liquid chromatography (HPLC) (1100-LC, Agilent Technologies). Aliquots of the drug suspensions were diluted (10x) in methanol in order to dissolve all BDP. The analyses were conducted with a mobile phase of methanol and water (3:1, v:v) at 25°C. BDP retention time was about 6 min and the wavelength of the detector was 242 nm. Chromatographic purity was calculated from relative ratio of the area under curve (AUC) of BDP peak and AUC of all peaks. For content assays, BDP was precisely weighted before treatment. Samples after laser fragmentation were entirely dissolved in methanol. The concentration of the samples was then measured by HPLC based on a concentration calibration line (R2 > 0.99). The content was then calculated as the ratio of measured concentration and theoretical concentration, normalized with the average ratio of suspension controls (as 100%). Samples for content assays were performed in duplicates.
2. 5. Physicochemical Properties
BDP suspensions were frozen at -80°C and dried under vacuum (< 100 x 10-3 bar) for 72 hours. BDP powders were then recovered from lyophilized samples for physicochemical characterization. The chemical composition was analyzed with Fourier transform infrared spectroscopy (FTIR) (FTS3000, Bio-Rad Laboratories) using the potassium bromide (KBr) pellet technique and with elemental analysis (EA) (ECS 4010, Costech). The cristallinity was evaluated with X-ray diffraction (XRD) (X'Pert X-ray, PANalytical Inc.) and with differential scanning calorimetry (DSC) (DSC7, Perkin Elmer).
2. 6. Jet Milling and Media Milling
For comparison with laser micronization, BDP powder was treated with a Fluid Energy jet mill (Telford, PA) at 86 psi (air), 86 psi (grinding) and 77 psi (pusher). For XRD comparison, a nanosuspension of BDP was prepared using a roller mill. A 30-mL glass bottle was charged with 10 mL of 0.8 mm diameter zirconium oxide beads, 150 mg of the drug and 7.5 mL of an aqueous solution of poloxamer 188 (0.8% w/v). The bottle was rolled at 70 rpm for 72 h. The media milled sample was washed by ultracentifugation (3 cycles of centrifugation at 17,000 x g for 20 min followed by redispersion in deionized water) to remove the excess of poloxamer.
3. Results and Discussion
We first conducted a screening study in order to obtain a preliminary assessment of BDP particle size and impurity after laser fragmentation. We irradiated BDP with femto- and nanosecond lasers and varied the fabrication parameters such as suspension concentration, volume, duration and laser power. Size, polydispersity index and impurity levels are presented in Table 1. We concluded from the screening study that higher irradiation power and duration produce smaller BDP particles, but at a cost of higher impurity. This conclusion was coherent with our previous studies on paclitaxel and megestrol acetate.
To further compare the size distribution and physicochemical properties with milled BDP particles, we selected four laser micronization conditions: (a) fs laser, 0.5 mg/mL, 10 mL, 400 mW, 1h; (b) fs laser, 2 mg/mL, 2mL, 400 mW, 30 min; (c) fs laser, 2 mg/mL, 10 mL, 50 mW, 16 h; (d) ns laser, 0.5 mg/mL, 10 mL, 5W, 1h. According to the screening study, BDP particles produced at these conditions were within the 0.5-5 µm size range with acceptable purity.
Table 1. BDP particle size (by DLS), polydispersity index (PDI) (by DLS) and chromatographic impurity (by HPLC) of BDP fragmented using fs and ns lasers under various conditions (n ≥ 3).
|
Condition |
Power/duration |
Average size (nm) |
PDI |
Impurity (%) |
|
Control suspension |
0 |
7460 ± 4680 |
0.7 ± 0.3 |
0.8 ± 0.2 |
|
fs, 0.5mg/mL, 2mL, |
50 mW 200 mW 400 mW |
1490 ± 170 1130 ± 300 1010 ± 200 |
0.3 ± 0.1 0.3 ± 0.1 0.2 ± 0.1 |
1.8 ± 0.1 2.2 ± 0.7 2.7 ± 0.5 |
|
fs, 0.5mg/mL, 10mL, |
50 mW 200 mW 400 mW |
1610 ± 200 1460 ± 240 990 ± 120 |
0.4 ± 0.2 0.4 ± 0.2 0.4 ± 0.1 |
1.0 ± 0.3 1.4 ± 0.1 2.1 ± 0.8 |
|
fs, 2 mg/mL, 2 mL, |
50 mW 200 mW 400 mW |
3380 ± 2630 2150 ± 690 1290 ± 260 |
0.4 ± 0.3 0.5 ± 0.3 0.4 ± 0.1 |
0.8 ± 0.1 0.9 ± 0.1 1.0 ± 0.1 |
|
fs, 2 mg/mL,10 mL, |
6 h 12 h 16 h |
1300 ± 300 1010 ± 310 880 ± 260 |
0.6 ± 0.2 0.6 ± 0.1 0.7 ± 0.2 |
1.0 ± 0.2 1.2 ± 0.3 1.6 ± 0.6 |
|
ns, 0.5 mg/mL, 10 mL, |
1.5 W 3.5 W 5 W |
7250 ± 2290 3230 ± 2590 2490 ± 1080 |
1.0 ± 0.1 0.9 ± 0.1 0.9 ± 0.1 |
1.1 ± 0.5 1.0 ± 0.4 1.2 ± 0.5 |
|
ns, 0.5 mg/mL, 10 mL, |
3.5 W 5 W |
2500 ± 820 1700 ± 720 |
0.7 ± 0.3 0.5 ± 0.3 |
1.5 ± 0.2 2.0 ± 0.6 |
3. 1. Size Distribution and Degradation
In order to confirm BDP particle size distribution after laser fragmentation, we evaluated one sample of each condition by laser diffraction (Table 2). Laser fragmented the BDP particle size to micrometric levels. The majority of laser-micronized particles were inside the 0.5-5 µm range. Laser-micronized particles were more dispersed than jet-milled particles, but longer duration of laser irradiation narrowed the particle size dispersion (see Table 2, the condition of ns laser 2h compared to 1h, other parameters are identical). The presence of large particles in laser-micronized samples was mainly due to sedimentation during laser fragmentation, as magnetic stirring did not generate enough agitation at the vial periphery.
Table 2. BDP particle size distribution (mean and cumulative undersize distribution at 10% (d10), median (d50) and 90% (d90)) measured by laser diffraction and relative content calculated from HPLC-measured / theoretical concentration ratio (mean ± standard deviation).
|
Condition (laser type, mg/mL, mL, hour) |
Size (µm) |
d10 (µm) |
d50 (µm) |
d90 (µm) |
Content (%) |
|
Control suspension |
10.8± 7.9 |
3.2 |
9.2 |
19.4 |
100 ± 4.7 |
|
Jet-milled |
1.5 ± 1.0 |
0.5 |
1.2 |
2.7 |
99.0 ± 0.1 |
|
fs, 0.5, 10, 1h (a) |
4.5 ± 5.4 |
0.6 |
2.6 |
10.6 |
93.6 ± 1.7 |
|
fs, 2, 2, 0.5h (b) |
5.1 ± 4.3 |
0.9 |
4.0 |
10.5 |
89.5 ± 3.1 |
|
fs, 2, 10, 16h (c) |
3.5 ± 4.8 |
0.5 |
1.8 |
8.9 |
95.8 ± 1.2 |
|
ns, 0.5, 10, 1h (d) |
4.2 ± 2.1 |
1.9 |
4.0 |
6.9 |
103.9 ± 2.5 |
|
ns, 0.5, 10, 2h |
2.4 ± 1.5 |
1.1 |
2.2 |
4.0 |
- |
Degradation of BDP evaluated by content assay is shown in Table 2. Content indicates the percentage by weight of BDP in the sample treated by laser fragmentation. Degradation induced by ns laser and jet milling was negligible, when standard deviation was taken into account. Fs laser induced slightly more degradation than ns laser and jet milling. However, in terms of efficiency and procedure simplicity, laser fragmentation was clearly advantageous. The output of laser fragmentation was nearly 100%, as the particles were directly irradiated in their glass container. As a comparison with jet milling, only 0.49 g milled particles out of the 2.07 g loaded BDP powder could be retrieved (yield 24%) due to the unavoidable loss of particles during the milling and of the residue in the equipment.
Untreated, jet-milled and laser-fragmented BDP particles were observed by SEM (Fig.1). Laser-micronized BDP particles had similar size and morphology as jet-milled particles. According to Fig.1, in most cases, laser-micronized BDP particles were within the 0.5-5 µm range. Larger particles may be present in less homogeneous suspensions, such as in condition (b) (Fig.1 bottom-left).
3. 2. Physicochemical Properties
In order to evaluate the chemical composition of BDP particles after treatment, we analysed untreated BDP powder, lyophilized control suspension, lyophilized laser-fragmented suspensions and dry jet-milled powder by Fourier transform infrared spectroscopy and by elemental analysis. FTIR spectrogram (Fig.2) showed no changes in the fingerprint region (500-1800 cm-1), but new peaks were observed at 3500-3600 cm-1 (OH band) in all BDP samples in aqueous suspension. This transition corresponded to the FTIR spectra of BDP monohydrate (Hunt and Patfield, 1989). BDP was converted to its monohydrated form (BDP•H2O) upon contact with water and this conversion occurred even in the absence of laser irradiation. Nevertheless, it is worth noting that both anhydrous BDP and BDP•H2O are commercialized anti-asthmatic drugs.






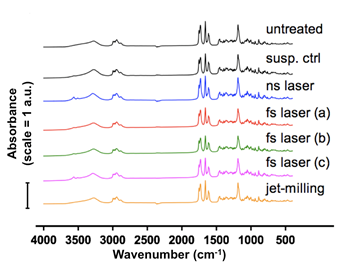
Elemental analysis (Table 3) was used to measure the content of carbon and hydrogen in BDP particles, and determined by subtraction the content of oxygen and chlorine (BDP formula is C28H37ClO7). As neither chlorine nor carbon was introduced into the sample, the increase of (O+Cl)/C ratio (Table 3) indicated an increase of oxygen in the samples. Again, this was mainly due to the formation of BDP•H2O in water. As a dry method, jet-milled BDP showed a similar profile with untreated powder. For BDP samples in contact with water, compared to the control suspension, laser-micronized BDP showed no obvious changes in its elemental composition. Thus, we concluded from FTIR and elemental analyses that the chemical properties of the laser-micronized drug were conserved.
Table 3. Elemental analysis of BDP particles. Oxygen is slightly more present in lyophilized BDP suspensions.
|
Condition (laser type, mg/mL, mL, hour) |
C (%) |
H (%) |
O+Cl (%) |
H/C |
(O+Cl)/C |
|
Untreated |
64.7 |
7.3 |
28.0 |
0.11 |
0.43 |
|
Control suspension |
63.0 |
7.2 |
29.8 |
0.11 |
0.47 |
|
Jet-milled dry powder |
64.4 |
7.2 |
28.5 |
0.11 |
0.44 |
|
fs, 0.5, 10, 1h (a) |
63.0 |
7.1 |
29.9 |
0.11 |
0.47 |
|
fs, 2, 2, 0.5h (b) |
62.5 |
7.0 |
30.5 |
0.11 |
0.49 |
|
fs, 2, 10, 16h (c) |
63.0 |
7.2 |
29.9 |
0.11 |
0.47 |
|
ns, 0.5, 10, 1h (d) |
62.5 |
7.1 |
30.4 |
0.11 |
0.49 |
In order to evaluate the physical structure of the BDP crystals, we conducted X-ray diffraction and differential scanning calorimetry analyses. The XRD spectrogram (Fig. 3) showed two groups with different spectra: 1) Dry powders that were never in contact with water – jet-milled BDP and untreated BDP – had identical spectra; 2) The spectra of laser-micronized BDP showed a mixture of original BDP and BDP•H2O. The peaks at 8° and 12° corresponded to BDP monohydrate (see referential XRD spectrogram of raw BDP and BDP•H2O in Valo et al., 2010). These peaks were more intense when BDP was immerged in water for a long time (fs laser condition (c) and ns laser condition (d)).
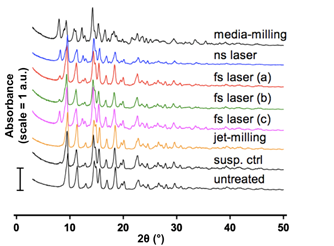
The spectra of BDP micronized by fs laser under conditions (a) and (b) were similar to that of the suspension control. Finally, the media-milled BDP sample showed a more pronounced presence of BDP•H2O. In summary, laser micronization did not induce changes in the crystalline phase of drug crystals. For drugs that are sensitive to hydration, their hydrate forms may appear in the water-suspension during laser fragmentation.
Table 4. DSC
analyses of BDP particles.
(Experiments
conducted in duplicates).
|
Condition (laser type, mg/mL, mL, hour) |
Melting point (°C) |
Fusion enthalpy (J/g) |
Relative enthalpy (%) |
|
Untreated powder |
210 ± 1 |
72 ± 17 |
100 |
|
Control suspension |
209 ± 1 |
63 ± 4 |
88 |
|
Jet-milled |
206 ± 1 |
62 ± 5 |
86 |
|
fs, 0.5, 10, 1h (a) |
198 ± 1 |
39 ± 4 |
54 |
|
fs, 2, 2, 0.5h (b) |
202 ± 1 |
53 ± 1 |
74 |
|
fs, 2, 10, 16h (c) |
203 ± 1 |
52 ± 7 |
72 |
|
ns, 0.5, 10, 1h (d) |
206 ± 1 |
56 ± 1 |
78 |
By using DSC, we measured the melting point and fusion enthalpy of BDP particles (Table 4). Relative enthalpy was calculated by taking the fusion enthalpy of untreated BDP powder as 100%. Both jet-milled and laser-micronized BDP particles had a lower melting point. Their enthalpy was also decreased and this decrease was more important for laser-fragmented BDP when compared to jet-milled BDP. The decrease in enthalpy could be caused by three factors: 1) As fragmented particles' surface/volume ratio was increased, less energy was needed for melting; 2) Impurity may be present in fragmented particles; 3) Part of the particles may be amorphous. Considering our previous conclusions that the crystalline phase was conserved, we propose that the loss in crytallinity was mainly caused by a partial amorphization of the surface of micronized particles and by the presence of chemical impurity in the samples.
4. Conclusion
We used femto- and nanosecond laser fragmentation to micronize a pulmonary drug model (BDP) as a proof of principle. Under different irradiating conditions, both lasers produced micronized particles of similar properties. Laser fragmentation required only minute amount (several milligrams) and had a higher output than standard milling methods. BDP particles were reduced to the size range of 0.5-5 µm with limited degradation. The laser process induced limited changes in chemical composition and in crystalline structure. However, a water-based process hydratation of sensitive compounds can be observed. Furthermore, laser fragmentation can cause amorphization at the surface of the particles.
To apply laser fragmentation to drug discovery, the technique could be further developed by enabling simultaneous irradiation of different compounds in a batch of wells or be optimized to suit specific pharmaceutical needs.
In conclusion, drug particles were successfully micronized by laser fragmentation with negligible degradation and physicochemical transformations. Laser fragmentation is thus a simple and efficient micronization tool, and is well suited for pharmaceutical R&D, especially for pulmonary drug discovery.
Acknowledgements
The authors acknowledge J.-C. Leroux and J. Leblond for helpful discussions and access to experimental equipments, E. Nadezhina for the elemental analyses and Y. Drolet, J.-M. Rabanel and X. Wang for technical assistance. Financial support was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Institutes of Health Research (CIHR), the Groupe de Recherche Universitaire sur le Médicament (GRUM), the Fonds de la recherche en santé du Quebec (FRSQ) and the Canadian Foundation for Innovation (CFI).
References
Asahi, T., Sugiyama, T., Masuhara, H. (2008) Laser fabrication and spectroscopy of organic nanoparticles. Accounts of Chemical Research, 41, 1790–1798. View Article
Besner, S., Meunier, M. (2010) Laser synthesis of nanomaterials, in "Laser Precision Microfabrication", Sugioka K., Meunier M. and Piqué A., Eds, Springer. View Article
Hobley, J., Nakamori, T., Kajimoto, S., Kasuya, M., Hatanaka, K., Fukumura, H., Nishio, S. (2007) Formation of 3, 4, 9, 10-perylenetetracarboxylicdianhydride nanoparticles with perylene and polyyne byproducts by 355 nm nanosecond pulsed laser ablation of microcrystal suspensions. Journal of Photochemistry and Photobiology A: Chemistry, 189, 105–113. View Article
Hunt, J., Patfield, J., (1989) Micronised beclomethasone dipropionate monohydrate compositions and methods of use. Sep. 12 1989, patent US 4,866,051. View Article
Jeon, H., Sugiyama, T., Masuhara, H., Asahi, T. (2007) Study on electrophoretic deposition of size-controlled quinacridone nanoparticles. The Journal of Physical Chemistry C, vol. 111, 14658–14663. View Article
Keck, C., Müller, R. (2006) Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. European Journal of Pharmaceutics and Biopharmaceutics, 62, 3–16. View Article
Kenth, S., Sylvestre, J.-P., Fuhrmann, K., Meunier, M., Leroux, J.-C. (2011) Fabrication of Paclitaxel Nanocrystals by Femtosecond Laser Ablation and Fragmentation. Journal of Pharmaceutical Sciences, 100, pp. 1022–1030. View Article
Merisko-Liversidge, E., Liversidge, G. G., Cooper, E. R. (2003) Nanosizing: a formulation approach for poorly-water-soluble compounds. European Journal of Pharmaceutical Sciences, 18, 113–120. View Article
Mosharraf, M., Nyström, C. (1995) The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. International Journal of Pharmaceutics.122, 35 – 47. View Article
Patil, P. P., Phase, D. M., Kulkarni, S. A., Ghaisas, S. V., Kulkarni, S. K., Kanetkar, S. M., Ogale, S. B., Bhide, V. G.(1987) Pulsed-laser-induced reactive quenching at liquid-solid interface : Aqueous oxidation of iron. Physical Review Letters, 58, 238–241. View Article
Rabinow, B. E. (2004) Nanosuspensions in drug delivery. Nature Review Drug Discovery, 3, 785–796. View Article
Rasenack, N., Hartenhauer, H., Muller, B. (2003) Microcrystals for dissolution rate enhancement of poorly water-soluble drugs. International Journal of Pharmaceutics, 254, 137-145. View Article
Sylvestre, J.-P., Tang, M.-C., Furtos, A, Leclair, G., Meunier, M., Leroux, J.-C. (2011) Nanonization of megestrol acetate by laser fragmentation in aqueous milieu. Journal of Controlled Release, 149, 273 – 280. View Article
Tamaki, Y., Asahi, T., Masuhara, H. (2002) Nanoparticle formation of vanadyl phthalocyanine by laser ablation of its crystalline powder in a poor solvent," The Journal of Physical Chemistry A, 106, 2135–2139. View Article
Taylor, K. (2007) Pulmonary drug delivery, in "Pharmaceutics: The Design and Manufacture of Medicines, 3rd ed", Aulton, M. E., Ed., Edinburgh : Churchill Livingstone
Valo, H., Laaksonen, P., Peltonen, L., Linder, M., Hirvonen, J., Laaksonen, T. (2010) Multifunctional hydrophobin: toward functional coatings for drug nanoparticles. ACS Nano, 4,1750–1758. View Article
Wagener, P., Barcikowski, S. (2010) Laser fragmentation of organic microparticles into colloidal nanoparticles in a free liquid jet. Applied Physics A: Materials Science & Processing, 101, 435-439. View Article